



The Arrow # 149

Hello friends.
A Unforced Error
I’ve got to start out this week by correcting an error I made in last week’s Arrow. A sharp-eyed reader (from France, of all places) emailed me that she couldn’t correlate what I had written with what was on the chart I showed. And she was correct. As I read what I had written, I had a difficult time figuring out how I even wrote it with the chart staring me in the face.
Here is the chart again, so you don’t have to go back and look for it.
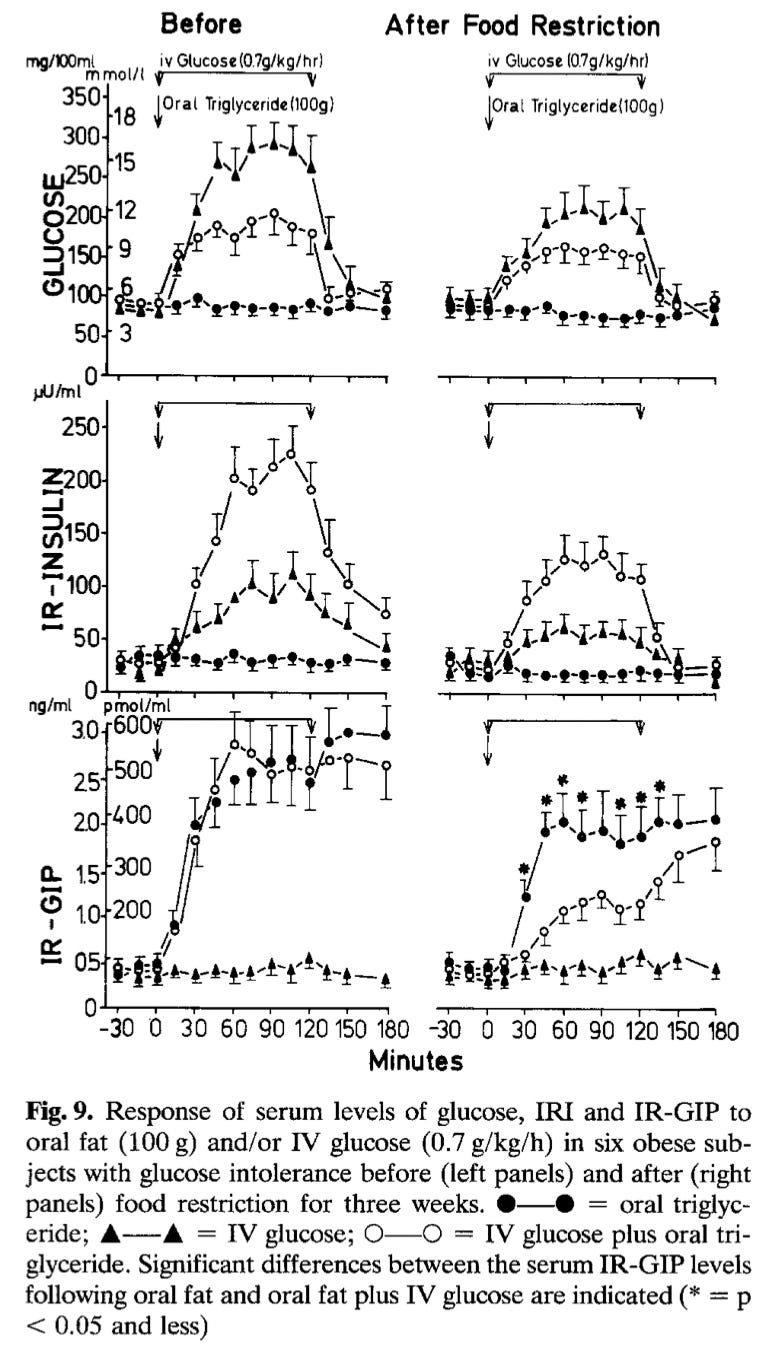
Take a look at the graph in the top left. As you can plainly see, the curve showing blood glucose as a function of just the IV glucose infusion is the highest of the three curves. It’s way up at the top. The middle curve, the one with the open circles, shows the glucose levels when dietary fat was added to the IV glucose. As any fool (except for me, apparently) can see, the blood glucose curve is lower than the curve generated by the IV glucose alone. And, the curve at the bottom showing dietary fat by itself generates almost no change in glucose.
Below is what I wrote about this last week. My email correspondent was kind enough to put in blue my incorrect interpretation.

I corrected my error and replaced the above with the following in the online version of the newsletter.
As you can see from the graph in the upper left, the oral fat drink didn’t do much to glucose, which is what you would expect from pure fat. An infusion of a fair amount of IV glucose does send the blood glucose curve to the moon. But when the oral fat is added, you can see that the glucose comes down, which makes the situation—at least blood sugar-wise—look a lot better.
What makes this so strange—to me, at least—is that the exact point I was trying to make was that if fat and carbs are consumed together, blood glucose levels don’t rise as much as they do if carbs are consumed alone. Which is exactly what the chart showed. Why I miswrote it is a mystery to me. But there it is.
I believe this is important because if you look at the next chart in the middle of the left side, you can see that insulin levels skyrocket when glucose and fat are combined. Which is a different picture than what you see in the blood glucose chart at the top left.
Fat by itself doesn’t do much in terms of raising insulin levels. Glucose by itself does provoke a rise in insulin. But the combination of fat and glucose sends insulin levels through the roof.
So many people are using continuous glucose monitors (CGM) these days and trying to eat in a way that minimizes blood sugar levels. Most people figure out pretty quickly that combining fat with carbs keeps glucose levels (as measured by their CGM) lower than eating carbs alone.
I discovered this myself, in fact.
MD and I both wore CGMs for a couple of months and tested all kinds of foods. Our typical evening meal is a chunk of some kind of meat (beef, if I have my way) along with asparagus (or something similar) and maybe three or four cherry tomatoes cut in half. We checked our blood sugar after this meal many, many times, and it showed almost no change in blood glucose levels for the next couple of hours.
One night I decided to try some ice cream just to see what happened. I consumed a small bowl—the equivalent of one scoop—and found that my blood sugar didn’t make a blip upward. Even after all the sugar in the ice cream. (It was Ben & Jerry’s Cherry Garcia, if you’re wondering.)
I couldn’t believe this. I figured the big steak I had eaten somehow kept my blood sugar in check despite the sugar the meal contained.
The next day I didn’t eat breakfast or lunch. Then, in mid-afternoon, I had another scoop of Cherry Garcia on an empty stomach. No moderating effect of any steak or anything else. My blood sugar didn’t do much of anything. After a scoop of ice cream! I was stunned.
MD’s comment was, “Well, you’re seeing only your blood sugar. You have no idea what your insulin is doing to keep it that way.” Then I remembered the papers I discussed in last week’s Arrow. Although—at this point, at least—there are no continuous insulin monitors to give me an insulin reading after I ate the ice cream, I suspect it was pretty high.
Which is the danger of tying to game CGMs. You can keep your glucose lower by covering your carb intake with fat (remember, the ice cream I ate contains plenty of both fat and sugar), but at the expense of your insulin levels. In my view, it’s a bad trade off.
The back and forth with the sharp-eyed reader who brought my error to my attention made me realize there is a lot about incretins, and GIP especially, that most people are unaware of, so I’ll spend a little time going over it.
But before I do, I’ve got to make an appeal.
To become a paid subscriber to The Arrow costs a measly 20 cents per day (19.78022 cents, to be exact, and that’s if you sign up monthly). If you sign up for an annual subscription, it’s only 16.438356 cents per day. A bargain to be sure. My single scoop of Cherry Garcia cost about five days worth of The Arrow. And The Arrow won’t raise your insulin.
More On Incretins
Last week we discussed glucose-dependent insulinotropic polypeptide (GIP) and how it stimulates a rapid insulin response when food comes in via the mouth and travels through the GI tract. Specialized cells, called K cells, are found in the inner lining of the upper small intestine. When these cells encounter carbs, protein, and/or fat coming down the line, they release GIP into the circulation, which signals to the pancreas to release insulin.
Insulin, as we all know, sends blood sugar levels down. It does this in a couple of ways. Most people think it drives it down by driving the sugar into the cells, primarily muscle cells. Which is true to some extent. But the main way insulin decreases blood sugar levels is by shutting off the liver’s blood sugar faucet.
What you may not realize is that your liver is almost constantly producing and releasing blood sugar. Right after a meal, especially a meal high in carbs, the liver more or less shuts off its production of glucose until the glucose from the incoming food is cleared from the blood, which the liver also helps with. But during most of the day and night while you’re not in the immediate post-meal phase (postprandial is the scientific term), your liver is cranking out sugar constantly to keep your blood sugar level stable.
The hormone glucagon is insulin’s opposite in almost all actions. Glucagon is called a counter-regulatory hormone to insulin. Like insulin, glucagon is made in the pancreas, but by a different type of cell. It is the accelerator to insulin’s brake pedal on the liver’s sugar production. Right after a meal, insulin will be high to store all the incoming sugar, fat, and protein away and glucagon will be scarce. A couple of hours after a meal, glucagon will be more abundant prompting the liver to produce some sugar to maintain blood sugar levels at an even keel. Insulin levels will be lower.
The above insulin-glucagon dance goes on constantly. It’s not something where one is turned on and the other off. It’s more of a wave-like form where both are present, but in varying amounts. At any given moment, the insulin to glucagon ratio determines what is happening at that point metabolically.
If asked, most doctors will tell you that of the two hormones, insulin is dominant. Which is not entirely true. One of insulin’s primary jobs is to regulate the release of glucagon, which, of the two hormones, is probably the more powerful in terms of the havoc it is able to wreak if unopposed.
Like insulin, glucagon acts through receptors called, logically enough, glucagon receptors. If you take mice that have been bred to have no glucagon receptors, they do just fine even with a normal amount of insulin. These mice make glucagon, but since they have no receptors, their glucagon doesn’t work. The signal can’t be heard. Their pancreases make insulin and their insulin receptors work perfectly, and they live pretty normal lives without any glucagon activity.
If you take normal mice—wild-type mice, in scientific lingo, ie, ones that can produce both insulin and glucagon and have working insulin and glucagon receptors—and give them streptozotocin (STZ), a drug that destroys their beta cells (the cells of the pancreas that produce insulin), these mice rapidly experience the onset of type 1 diabetes. They go into ketoacidosis and die fairly quickly.
However, if instead you destroy the beta cells of the glucagon-receptor knock-out mice with STZ nothing much happens. They continue to mouse along pretty normally. Even without any insulin. These mice now can’t make insulin and have no ability to detect the glucagon signal. In effect they have neither.
Roger Unger probably did more research on this over the years than anyone. I clipped the segment below from a longer, technical lecture of his. In this short video, he touches on a few issues he has with insulin versus glucagon. Insulin was discovered before glucagon, and its use in 1932 to save people doomed to die from type 1 diabetes was viewed as miraculous. And without a doubt it was for those doomed people. Consequently, we have lived in an insulincentric world since. The video shows two mice in a divided box. Dr. Unger says the mouse on the left is very sick. He misspoke or the image might be reversed. At any rate, he meant the mouse on the right from our perspective, i.e., the one with all the wet sawdust in its cage as a result of its constant urination.
Here is the situation with these two mice. Both of them have had their beta cells (the insulin makers) destroyed by STZ. The mouse on the left is getting a drug that suppresses glucagon, so it isn’t really getting any kind of glucagon action. The sick one on the right is getting a saline solution, so its glucagon is working at full capacity.
You can clearly see the difference.
In the last part of the video, glucagon-receptor-knockout-mice that have had their beta cells wiped out by STZ have normal glucose levels. But when they are given temporary glucagon “receptors,” their blood levels of glucose skyrocket. After the temporary receptors wear off, so to speak, their sugar levels return to normal.
Having watched this short video, you should be impressed by the potency of glucagon. Now that you know what glucagon does, we can go into some of the other effects of GIP.
So far, we’ve discussed how GIP signals to the pancreas to increase insulin levels. It also calls for a release of glucagon as well. Which seems kind of counter productive, but it really isn’t.
It was hard to tell from the charts I showed last week, but dietary fat all alone will stimulate the release of GIP. This release of GIP tells the pancreas that food is coming down the pike, so squirt out some insulin to keep the blood sugar from going too high. But in the case of nothing but dietary fat, there is no blood sugar rise, because there isn’t any sugar in the fat. So this little squirt of insulin will then drop the blood sugar maybe too low.
Which is why GIP also calls for glucagon. The glucagon ensures that the blood sugar doesn’t drop too low as a consequence of the insulin.
It all seems kind of counterproductive. But I haven’t told you the full story yet.
GIP does a number of other things besides just calling for insulin and glucagon.
Here is a graphic showing some of them.

The one down at the bottom is extremely important. GIP is involved in increasing bone formation and decreasing bone resorption. Although it doesn’t look like it when you see a skeleton, bone is an active organ. It is constantly dissolving and being reformed. This is called bone remodeling, and it goes on all the time. If certain bones end up carrying a lot of weight from a particular direction, the part of the bone that needs reinforcement gets it while the part that isn’t carrying as much load, gets smaller. The skeletal structure is under constant remodel to ensure a good underlying framework.
And GIP plays a fairly major role in this. So much so, that in people who for whatever reason have to get their nutrition via intravenous feeding for any length of time end up having a lot of bone loss. Since their food isn’t coming in through the GI tract, there is no GIP produced and thus no GIP effect on bone remodeling. So the bones mainly dissolve. It’s a real problem.
So, if we eat a diet with a lot of protein and fat and not a lot of carbs—a diet similar to the one we evolved on—GIP is stimulated so we can still build our bones (and have plenty of protein to help do it). But since neither fat nor protein drive blood sugar up, GIP needs to drive the release of a bit of glucagon along with the insulin to keep our blood sugar stable as we remodel our bones.
Ain’t nature grand?
Oh, and there is another incretin you may have heard of. It is glucagon-like peptide-1 (GLP-1). It is the much more famous cousin of GIP. Like insulin and glucagon, GLP-1 has a receptor. Drugs that block receptors are called antagonists. Many drugs are in development that are glucagon receptor antagonists. If we could somehow block the action of glucagon, we could treat diabetics much more effectively than with the use of insulin. As far as I know, none of these glucagon receptor antagonists have been approved yet.
Drugs that stimulate a particular receptor are called agonists. There are a number of drugs currently on the market that are agonists for the GLP-1 receptor. One of the most famous is semaglutide, sold under the trade name Ozempic and Wegovy. As I wrote above, GLP-1 is GIP’s much more famous and profitable cousin.
Since the discovery of incretins, researchers have been looking at them for possible diabetes and weight-loss drugs. They struck gold with GLP-1, but haven’t had much luck with GIP. So far they haven’t figured a way to make it into a drug, so there hasn’t been nearly as much research on GIP as there has been on GLP-1. But that is going to change.
Just yesterday the FDA approved tirzepatide, trade name Mounjaro, as an anti-obesity drug. Mounjaro was the name when the drug was approved for use in diabetics, but the same drug, now approved for obesity, will be called Zepbound. (It floors me sometimes that they pay people big salaries to come up with catchy names for drugs and get something like Mounjaro or Zepbound for their coin.)
Zepbound, like Ozempic and Wegovy, is a GLP-1 agonist. But in addition, it is a GIP agonist as well. It stimulates receptors to both. Since it has shown the best results in terms of weight loss, I’m sure its success will prompt a lot of researchers to look more closely at GIP. I suspect over the next few years, we’ll find out a lot more about GIP than we know now.
Nothing like the potential for a multi-billion-dollar drug to fire up the research community.
Resignation of Stanford’s President

Not long ago there was a major brouhaha over the resignation of Stanford University’s president Marc Tessier-Lavigne. He was accused of academic fraud for publishing scientific articles filled with false data.
As it turns out, that wasn’t exactly the case. He was basically run off because of his commitment to free speech and his apology to a federal judge who had been shouted down during a law school presentation.
The Wall Street Journal has all the details in a must-read article. Here it is unpaywalled. After seeing what happened to Tessier-Lavigne, it’s easy to see why professors would be afraid to speak out. And why major university campuses are hotbeds of wokeism and far left ideology and suppression of free thought and speech.
Coronary Calcium Score Calculation
One of the most common questions I get asked via email is about the different scoring technique for coronary calcium scans I wrote about a couple of years ago. Since I wrote that particular issue of The Arrow on another platform that is no longer available since I switched to Substack, folks can’t go back and search for it. I have copies of these old Arrows from years back, but I don’t have them in a way I can make accessible right now, but I’m working on it.
Instead of waiting for that time, which, given my tendency to procrastinate and time’s to fly, may not come to pass for another couple of years, I’ll reprint what I wrote here. Where it will be searchable. What follows will be the combination of two different Arrow segments on the same subject, with a little editing thrown in for clarity. Since there will be some editing, and since I was the writer, I’m not going to put it all in quotes.
The first post I wrote was in answer to an email question about coronary calcium scanning (CAC, which stands for Coronary Artery Calcium).
I'm uniquely qualified to answer this question, because I used to be a part owner of a scanning facility. We did not only coronary artery calcium scans (CAC), but all kinds of other scans as well. Virtual colonoscopies, scans to look for osteoporosis, liver fat, early lung cancer and all kinds of issues. When I got involved, as is my wont, I immersed myself in the literature.
CAC screening is the ONLY way to actually visualize any degree of coronary artery disease non-invasively. You can see narrowing of coronary arteries if you do a coronary angiogram, but that is far from non-invasive. All the other ways people use to determine coronary artery disease are by using purported surrogate lab values such as LDL-cholesterol levels, which are absolutely worthless.
A CAC scan uses technology that rapidly fires either electrons or X-rays through the heart area. A regular chest X-ray is too slow as the heart beats fast enough to blur any image of individual coronary arteries. Expensive equipment--vastly more expensive than a standard X-ray setup--fires either electrons in the case of an electron beam tomography (EBT) scanner (the first, and still the best in my view) or a specialized rotating CT scanner (that takes a lot longer and gives the patient a lot more radiation exposure).
The end result is a picture of the heart and the coronary arteries that supply the blood needed to keep it beating.
Any plaque present in the coronary arteries contains some calcium. Unlike the soft tissue in the body, calcium shows up on an X-ray. Think of bones. They contain a lot of calcium and so really stand out white against the black film when you see an X-ray of some part of your body. The same holds with calcium in the plaque in coronary arteries.
A cardiologist named Arthur Agatston, M.D. developed the scoring system used to give an objective number to CAC results. (You may know Dr. Agatston as the developer of the South Beach Diet, a low-carb diet book popular a decade or so ago.) The Agatston score is basically a measure of how much calcium is in any given coronary artery. And it is dependent upon the density of the calcium that shows up in the scan.
The more calcium picked up by the scan converts to a score that supposedly determines one's heart disease risk. Which makes sense, because it is, after all, a direct visualization of plaque.
In my view, the CAC is an excellent way to determine the extent of plaque someone is burdened with. Which is the way most docs see it. If you've got a high calcium score, you've got a lot of plaque, ergo, you are at great risk for having a heart attack.
But it's not that cut and dried. I learned that CAC as currently calculated wasn't a true measure of the risk for heart attack. Way back in the day when I was working on the paleopathology chapter in Protein Power, I came across an article by George V. Mann, who had done many autopsies on the members of the Masai, nomadic herders in Africa who follow a diet of primarily meat, milk, and blood. In studying the Masai, Mann found that they had almost no incidence of heart disease or heart attacks. But autopsies showed they had a ton of calcium in their coronary arteries that were filled with plaque. (If you are interested, here is my Dropbox link to this Mann study.)
As it turns out, calcium stabilizes plaque. There are two types of plaque: stable and unstable. Unstable plaque is the plaque that is prone to rupture, develop a clot, and cause a heart attack. Stable plaque is, well, stable. Seems to cause very little, if any, problems.
The way Dr. Agatston derived his equation for scoring the calcium in coronary arteries weighed heavily in favor of the density of the calcium in the plaque. But, as it turns out, there is more to it than just calcium. There is also a plaque volume that's reported on in a CAC scan. Most people ignore the plaque volume number and focus on the CAC, or Agatston, score.
If a person has a large volume of plaque, but very little calcium, that means the plaque may well be unstable. And prone to rupture, form a clot, and lead to a heart attack. So it's important to take the plaque volume into consideration.
Let me expand on this a bit. Most people seem to believe that heart attacks come about when the plaque lining a coronary artery becomes enlarged to the point that it shuts off the flow of blood. This isn’t really the case. What happens is that the unstable plaque ruptures, releasing its contents into the artery. The body’s healing response then kicks in and sends all kinds of clotting factors to the rupture where a clot forms. This clot can then break free and ride the bloodstream in the artery down to a narrower location, where it then occludes the artery. The heart muscle nourished by the artery downstream from this clot then suffers from a loss of oxygenated blood and all the familiar symptoms of a heart attack follow.
Which is why unstable plaque is so dangerous vis a vis stable plaque. The kind George Mann found in the Masaii. Stable plaque doesn’t rupture.
A paper came out in 2014, which was right in the middle of my time as a part owner of a scanning center. One of the authors of this paper is a guy named Matt Budoff, who has been involved with CAC since its earliest days. He's written a number of papers on CAC, and knows as much as anyone does about the process. Which made me feel much more comfortable about the work that went into the paper.
The authors of this paper (also a Dropbox link) looked at a different way to stratify people who had positive CAC scans. Their formula involves both the volume of plaque and the density. Problem is, the readings that show up on a CAC report typically list the plaque area and the density. The report is taken from a two-dimensional scan, and volume is a three-dimensional number. In order to calculate the volume, you've got to have another dimension, which you can get from the folks who run the scanner. Most scanners run either a 2.5 mm slice or a 3 mm slice, so you just have to find out the slice size, and you can calculate volume.
Once you have the volume and the score, you can generate another number, which puts you into a risk category. The risk category has been determined by following countless patients with known scores and seeing how many actually had heart attacks.
Once I started running these new risk numbers, I discovered that the vast majority of subjects who had high CAC scores--even scores up in the 500s and 600s--had minimal risk.
And I learned something else important.
Many people who went on low-carb diets reduced their plaque volume. And in doing so, increased their calcium density. Their CAC scores went up a bit, but their risk as stratified by the Budoff paper went down. Their plaque had stabilized.
Here is how to do the calculations. Let me show you a typical calcium scan readout. This one came from a friend of mine in Colorado. Different scanning centers have different ways of presenting their scans, but most of them include a plaque volume and a density score (the Agatston score).
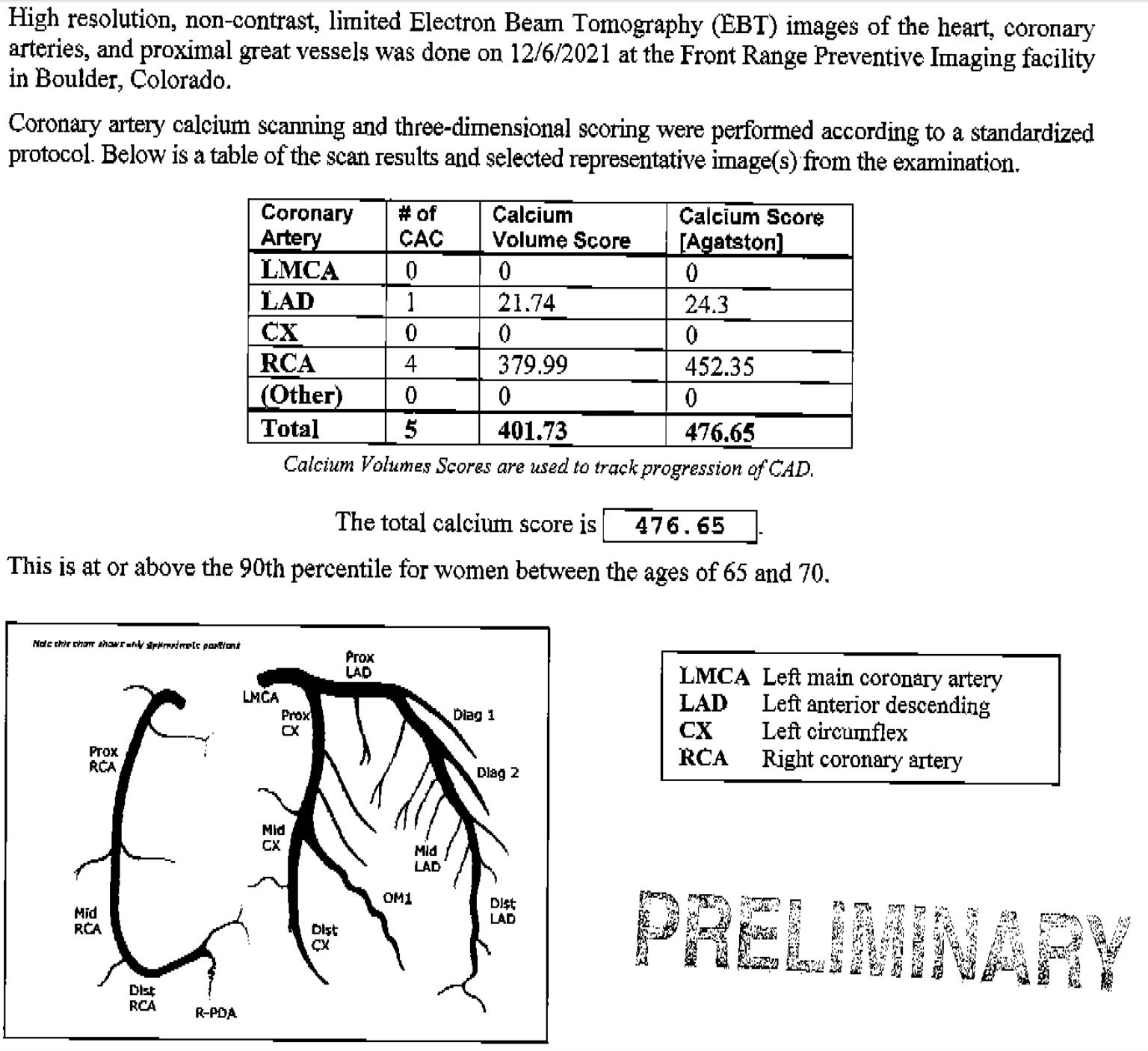
In the readout, you can see the individual scores for each coronary artery along with the combined score for all. What this scan doesn't show is the slice size the machine uses. Most scans don't show that, so you'll have to call the imaging center to get that info, which they should be more than happy to provide. It's going to be either 2.5mm or 3mm. In this case, it was a 3mm slice.
Once you have that in hand along with your scan, you're ready to roll. Here are the steps.
divide the volume score by the slice thickness
take that number and divide it into the total calcium score
look up that number on the chart below
That's pretty much it. Let's walk through the score I posted above.
The volume is 401.73. Divide that by 3mm (3). 401.73/3=133.91
Divide the total calcium score by that number. Total score in the scan above is 476.65. 476.65/133.91=3.56
You then see where that number fits in the quartiles on Table 2 on page 274 of the article:

I've put a red box around it in the graphic above. The higher the quartile, the lower the risk.
In the scan we calculated above, the 3.56 fits comfortably in the 4th quartile, which represents low risk. So this patient has a low risk of a heart attack despite having a high calcium score.
Here I show the risk as measured on the graphic from page 276 of the paper:

If all this is of interest, I would encourage you to read the brief introduction in the paper I linked above. It will explain why this method is superior to the standard Agatston calcium score.
Finally, these calculations work only if the total calcium score is 130 or higher. The authors of the paper write:
To qualify as a calcified plaque using CAC scoring, the plaque calcium density, measured in Hounsfield units, must be 130 Hu or higher
I hope this clears everything up on this method of determining risk based on CAC scoring.
More on the Keto vs the SuK Diet
I ran out of time last week, so was only able to cover a tiny portion of the outcome of this important study.
To recap, the researchers recruited ten healthy, lean (BMI ~20.5), pre-menopausal females (avg age ~32) who had been on ketogenic diets for an average of about four years. These women first maintained their typical ketogenic diet for the first phase of the study lasting 21 days. During this time, they were tested for a number of biomarkers for aging and health.
During the second phase, another 21 days, the subjects consumed the standard UK diet (SuK), which recommends the “consumption of at least 267 g of carbohydrate per day for women.” In the study, the researchers call this diet the standard UK diet (SUK) and the suppressed ketosis diet (SuK). I prefer Suk since it is closer to sucks, which better describes the diet, at least in my view, although there is no doubt it did suppress ketosis.
On the third 21 day phase of the diet, the subjects returned to their previous ketogenic diet.
During the study, daily measurements of beta-hydroxybutyrate (BHB) were taken and “ageing biomarkers and anthropometrics were evaluated at the end of each phase.”
Here are some of the findings.
First, last week I showed the difference in ketosis during the three phases.

This graphic showing what happened to BHB during the phases leaves little doubt that ketosis was suppressed by SuK.
How about glucose levels?

As you might predict, they went up a little during phase 2. But you’ve got to remember that these are lean, healthy young women, so you wouldn’t expect a huge rise. But look at the amount of insulin they had to release to keep their blood sugar from shooting too high.

Pretty impressive change. Ketosis suppression significantly increased. Insulin went up 1.78-fold, almost double during the SuK phase. Don’t let anyone tell you that a high-carb diet won’t run your insulin levels up.
Also in the SuK phase of the diet, subjects saw their IGF-1 levels rise even a bit more than their insulin levels did.

As the authors explain
Chronically elevated IGF-1, and/or increased IGF-1 bioavailability and sensitivity, receptor expression, and amount of Ras protein prenylation are strongly implicated in neoplasia [cancer] and ageing, whilst IGF-1 knockdown within in vivo models show improved longevity.
In observational studies, low levels of insulin and IGF-1 have also been associated with reduced levels of pathologies. For example, elevated IGF-1 has been shown to correspond to a 69% increase in colorectal cancer risk, a 49% increase in prostate cancer risk, 65% increase in breast cancer risk, and a 106% increase in lung cancer risk (relative risks). Notably, a recent meta-analysis involving over 30,000 participants indicated that IGF-1 within the range of 120–160 ng/mL was the optimum range associated with the lowest risk of all-cause mortality. The participants in the present study fell well within this range during the P1 and P3 phase; however, during SuK (P2), IGF-1 significantly increased, which may confer an increased risk of all-cause mortality. Conversely, the lower levels of insulin and IGF-1 during the P1 and P3 phases may be of health benefit given that higher levels of IGF-1 and insulin are significant risk factors for various diseases.
These results discussed in the quoted paragraph above all come from observational studies, so they don’t prove causality. But I would certainly rather have my IGF-1 in the optimal range as described than way above it.
You can see that thyroid hormone goes up when carbs go up. And goes down when carbs go down. I discussed this phenomenon as it applies to thyroid hormone and low-carb diets in an earlier Arrow.

If you look at inflammatory markers in general, you can see they all went up on the SuK diet and down upon return to the ketogenic diet.

The same with markers of inflammation in the liver.

Below is the chart of what happened on the oral glucose tolerance test (OGT) taken at the end of each phase of the study.

Let’s walk through it starting with the OGT at the top.
You’ve probably heard that if you’ve been on a low-carb diet, you should up your carbs for a few days before taking an OGT. From the OGT shown above, you can see why. The subjects on the first phase of the ketogenic diet showed a larger increase in the glucose curve as compared to when they were at the end of their 21 day SuK diet. After they went back on their ketogenic diet, their blood sugar levels were again higher than when on the SuK diet. And you can see on the tail end of the SuK diet that the blood sugar started creeping back up at the 180 minute point. It did not do that on either of the ketogenic phases.
From the second graph, you can see that insulin went higher during the OGT while the subjects were on the SuK phase.
And, finally, ketosis was completely inhibited during the SuK phase whereas during the two ketogenic diet phases the oral glucose load knocked the subjects out of ketosis, but over a fairly short period of time, the BHB levels rose back up. Confirming what I wrote last week about carb adaptation and how it doesn’t go away after a carb blowout. These subjects were on a high carb diet for three weeks and quickly got back into their low-carb adapted state.
Some caveats about this study.
First and foremost is that these are young, healthy, slender women. If you are obese and out of shape, these findings from the SuK phase of the diet would probably be of much greater magnitude. See the very first graph at the start of this Arrow, the one I made the mistake on. If you look at the charts on the right hand side of the graphic, you can see they are not as extreme as those on the left. But when the measurements were taken for the charts on the right, the subjects had fasted for three weeks and lost over 20 pounds.
If you are older, your results may vary. If you are male your results may vary. But, based on the findings of this study, I would confidently predict that if you gave up a SuK diet and went on a good quality ketogenic diet, just about all your markers for insulin, inflammation, and aging would improve.
Also, these subjects have been on the ketogenic diet for an average of ~4 years. As you can see from how quickly they bounced back from their 21 day SuK diet, they have excellent metabolic flexibility (a subject I promise to write about in more detail soon). If you are overweight and have glucose intolerance or any of the other components of the metabolic syndrome, it’s doubtful that you could go on a ketogenic diet for a couple of weeks and normalize your lack of metabolic flexibility. It will improve. That I can just about guarantee. But it will take a while adhering to the plan to completely normalize, if it ever does.
Video of the Week
Okay, this video will take a little explanation.
One of my intellectual heroes is the economist and philosopher Thomas Sowell. He is finally getting his intellectual due as even some left-wing academics are finally recognizing his genius. Sowell started his academic life as a Marxist and ended up as a conservative, which doubtless cost him in terms of fame and all the prizes academics shower themselves and each other with.
If I could have a selfie taken with anyone in the world, I would probably choose Thomas Sowell. I’m ashamed to admit that I have a friend who works with him at the Hoover Institute at Stanford and have never taken him up on his offer to introduce us. I just haven’t been anywhere near Stanford in years. And Sowell is now 93, so the clock is ticking. I need to just make the effort.
Here is a great video of Sowell squaring off against a feminist in the early 1980s. The first thing you’ll note is how different feminists from the early 1980s look as compared to feminists today. I love how this one is able to maintain her stoic and supercilious nature as Sowell destroys her arguments. His arguments are basically the same as those of this woman who recently won the Nobel Prize.
I’ve got the video queued, but the whole thing is interesting to watch.
Enjoy!
It’s time for a poll.
Even if you hate it, click the like button just to give me a false sense of security.
Okay, that’s it for this week. This one wasn’t of Tolstoyian length like the last one, so you probably whipped through it. Keep in good cheer, and I’ll be back next Thursday. Probably from somewhere in the wilds of Arkansas. We’re heading there next week to get together with the fam to celebrate MD’s birthday.
Thanks for reading all the way to the end. Really, thanks, especially on this one. If you got something out of it, please consider becoming a paid subscriber. I would really appreciate it.
Finally, don’t forget to take a look at what our kind sponsors have to offer. Dry Farm Wines, HLTH Code, Precision Health Reports, The Hustle (free), and now The Morning Brew (also free).
Sowell is also one of my intellectual heroes. I remember how sad I was when he announced he was no longer going to write his Forbes column because of his age. (When I was younger it didn't occur to me that my heroes would grow old and infirm.) But years later he is still writing books and doing interviews (with Peter Robinson in his Uncommon Knowledge podcasts), his mind still sharp at 93. It gives one hope that we won't all end up like Joe Biden.
I often times struggle to distinguish some of the information in the graphs and end up glossing over them but try to get the gist of what you are explaining. So I appreciate when you summarize the point.
I watched both videos through this time which I don’t always do, I know you can tell how many people are clicking on the links. I did immensely enjoy the Firing Line one as Dr. Sowell holds his own and knows his data to back up his points as this lady seems to want to control the direction of their discussion.